Nature Humaine (amocalypse)
Théorie>Electricité>Electrochimie
Première version: 21/04/2001
Dernière version: 2010-11-22
L'électrochimie
Sommaire de la page
Préambule
Attention! Dans ce cours, comme pour celui de l'électricité,
j'inverse le signe des charges, à savoir que l'électron est de charge
positive et le proton de charge négative (à l'inverse des conventions
électriques de 1800, toujours appliquées aujourd'hui). Le courant électrique
électrique a donc le même sens que le courant d'électrons.
Cela change juste le signe des bornes de la pile (c'est plus facile à
comprendre comme ça), et e- devient e+.
1) Définitions préalables
Une réaction électrochimique est une réaction où à lieu un échange d'électrons (e+) entre un réducteur et un oxydant.
Ces réactions ont lieu dans une cellule (pile électro-chimique ou électrolyseur), qui consiste en un électrolyte contenant les ions, faisant office de conducteur ionique pour permettre aux électrons d'aller d'une électrode à l'autre, et des électrodes où ont lieu les réactions électrochimiques.
La cellule peut avoir 2 fonctionnements possibles :
- pile (le potentiel appliqué sur le circuit extérieur à la
cellule étant inférieur à la fem de la cellule, c'est elle qui fournit le
courant, l'anode (lieu de l'oxydation=> perte d'électron) est donc la borne
+ de la pile (fournissant les électrons issus de l'oxydation au circuit)).
- électrolyseur (le potentiel appliqué aux électrodes
étant supérieur à la fem de la cellule, elle se contente de faire passer le
courant entre ses bornes, l'anode étant sur l'électrode reliée au "-" du
circuit, là où arrivent les électrons qui ont parcourus le circuit, ces
électrons engendrant une réduction des molécules précédemment oxydées,
donc redonnant les électrons précédemment empruntés), la cellule se
comportant comme une résistance + une fcem.
Fonctionnement en pile
Nous considérons en premier temps le fonctionnement en pile de la cellule, quand il n'y a pas de courant appliqué aux électrodes pour produire une électrolyse mais que le système est laissé libre (électrodes reliées entre elles à l'extérieur de l'électrolyte par un voltmètre ou un circuit électrique sans f.e.m). Les réactions aux électrodes sont donc celles qui vont avoir lieu spontanément.
Définitions de base
- Le potentiel d'une réaction est l'énergie (exprimée en Volts)
fournie par la réaction. Si on faisait tomber un électron d'une ddp
valant ce potentiel, on récupérerait la même énergie.
- Le potentiel d'une réaction peut aussi être vu comme la "volonté" qu'a la
réaction pour s'effectuer. On peut ainsi mesurer si une réaction se fera au
détriment d'une autre en sachant si cette réaction aura plus de force que
l'autre pour s'effectuer.
- V° = potentiel standard d'une réaction se déroulant dans les
conditions standards (T=298K=25°C et pH=0).
- Veq = potentiel à l'équilibre thermodynamique.
- [espèce] indique la concentration molaire (proportionnelle à
l'activité) de l'espèce dans la solution (en mole/litre). Si dans
l'équation d'oxydation, une espèce est présente n fois (ex:
2H2O) alors l'exposant de sa concentration est n
(ex: [H2O]2). La concentration d'un précipité
(donc d'un solide) est toujours égale à 1.
- p devant une constante (H par exemple, symbolisant l'activité de l'ion
H+ en solution, ou encore la concentration des ions
H3O+ dans la solution) signifie pH = - log H. Autre
exemple, pKe = - log Ke).
- rappels de calculs avec les logarithmes: log (ab) = log a + log b ; log (a/b)
= log a - log b ; log (ax) = x log a ; log A = - pA (notation pour
simplifier l'écriture).
- La valence n d'un atome est le nombre d'électrons qu'il perd
lorsqu'il s'ionise. Ce nombre peut changer suivant la réaction, le fer pouvant
perdre entre 1 et 3 électrons suivant la réaction considérée.
Soient les deux demi-réactions suivantes:
* A l'anode (+) : oxydation: Red1 ⇌ Ox1 + n1 e+
avec Ox1 et Red1 le couple oxydo/réducteur du même élément.
VRed1/Ox1 = potentiel de l'oxydation = -VOx1/Red1
* A la cathode (-) : réduction: Ox2 + n2 e- ⇌ Red2
VOx2/Red2 = potentiel de la réduction
Les potentiels sont généralement donnés dans le sens de la réduction: VOx/Red.
Un oxydant (ou accepteur) prend des e+, donc se réduit. Un réducteur (ou donneur) donne des e+, donc s'oxyde. Un oxydé est donc un réducteur et un réduit est un oxydant...
Les deux demies réactions donnent la réaction d'oxydoréduction (ou globale) suivante:
n1 Ox2 + n2 Red1 ⇌ n1 Red2 + n2 Ox1
avec Red1/Ox1 appelé le premier couple redox, et Red2/Ox2 le deuxième couple redox. Le sens 1 (de gauche à droite), et favorisée si VOx2/Red2 supérieur à VOx1/Red1. Sinon, c'est le sens 2 (de droite à gauche) qui s'effectuera spontanément. La réaction est d'autant plus rapide que l'écart entre les deux potentiels est important. A noter que chaque demi-réaction est multipliée par le nombre d'e+ que nécessite l'autre demi-réaction, afin que la réaction globale soit équilibrée.
Le potentiel de la réaction d'oxydoréduction est donné par:
Vfem = Δ(VOx2/Red2 ; VRed1/Ox1 ) =
différence de potentiel des 2 demi-réactions (noter que pour
la réaction d'oxydation, on prend le potentiel de l'oxydation, donc de signe
contraire au potentiel de réduction donné par la table). C'est la
force électromotrice de la cellule.
Donc, pour une cellule électrochimique de type Red1/Ox1 // Ox2/Red2, (donc
VOx2/Red2 supérieur à VOx1/Red1, la réaction se fait
dans le sens 1) son potentiel est donné par:
Vfem = VRed2/Red1 = VOx2/Red2 - VOx1/Red1= Veq/sup - Veq/inf
La réaction d'oxydoréduction est spontanée si Vfem > 0 ou si ΔG<0.
ΔG = - n F Vfem
avec n le nombre d'e+ impliqué dans la réaction en tant qu'e+ libres (n1*n2), c'est la valence.
F la constante de Faraday (en C), c'est la quantité d'électricité nécessaire pour électrolyser 1 mole de matière
ayant une valence de 1.
G l'enthalpie libre de GIBBS (en J) telle que ΔG°= ΔH° - TΔS°
L'enthalpie libre G peut être vue comme l'énergie minimale à fournir pour casser ou former une liaison (assimilable à
l'énergie de liaison des molécules, en J/mol). C'est l'énergie fournie par une quantité d'électricité n.F (en C) pour
passer d'un potentiel de Vfem à un potentiel arbitraire de 0 V.
Si les activités de toutes les espèces sont unitaires, les G et V de la relation précédentes sont remplacées par G° et V° (valeurs standards).
Le potentiel d'une demi-réaction (dit aussi potentiel d'électrode), lorsque les conditions ne sont pas standards, est donné par la loi de Nerst (dans les conventions internationales [Red]/[Ox]) :
VOx/Red = V°Ox/Red - (RT/nF) ln (aRed/aOx)
Cette équation se simplifie à T=25°C = 298 K : on multiplie tout d'abord RT/nF par 2,303 pour passer du ln au log, et en remplaçant les constantes R et F par leurs valeurs, puis la température T par 298 K, on transforme (RT/nF) ln par (0,06/n) log, ce qui nous donne comme équation:
VOx/Red = V°Ox/Red - (0,06 / n) log (aRed/aOx)
Activité: C'est la capacité des espèces à se déplacer dans le milieu dans lequel elles se trouvent. L'activité est proportionnelle à la concentration de l'espèce: aRed = γ[Red]. Dans un solide, γ=0, et dans un liquide, γ=1. Donc on peut assimiler l'activité à la concentration.
Equation de Nerst pour l'équation globale:
Vfem = V°Ox2/Red2 - V°Ox1/Red1 - (RT/F) ln ([Ox1]n2 [Red2]n1)/ ([Ox2]n1 [Red1]n2)
Avec V°Ox2/Red2 - V°Ox1/Red1 le potentiel standard de la réaction. Donc aRed et aOx se résument pour aRed à la concentration de chaque espèce à droite de l'équation d'oxydoréduction (et non seulement de la demie réaction) et pour aOx à la concentration de chaque espèce à gauche. Ex: aOx = [Ox2]n1 [Red1]n2.
([Ox1]n2[Red2]n1)/ ([Ox2]n1[Red1]n2) peut se mettre sous la forme d'une constante K, appelée constante d'équilibre de la réaction.
K = e (-ΔG° / RT)
K doit être supérieur à 104 pour que la réaction dans le sens 1 soit suffisamment rapide au niveau industriel, et inférieur à 10-4 pour la réaction rapide dans le sens 2.
Définition de la variation d'énergie libre: ΔG = - n.F.V, avec n le nombre d'e+ impliqués dans la réaction considérée, et V le potentiel du couple rédox de la réaction. Les variations d'enthalpie libre pour les demi-réactions sont ΔG1 = - n1.F.VOx1/Red1 et ΔG2 = - n2.F.VOx2/Red2. Pour la réaction globale, ΔG = n1.ΔG2 - n2.ΔG1 = - n1n2.F(VOx2/Red2- VOx1/Red1) (à noter que l'on multiplie par le nombre d'atome de l'autre demi-réaction). Pour la réaction globale, si les conditions ne sont pas standards, on peut retrouver ΔG = ΔG° + RT ln K. Donc:
ΔG° = - RT ln K = - n F Vfem
et avec une température de 25°C: log K = n Vfem / 0,06
Etat d'oxydation
Etat d'oxydation : C'est le nombre d'e+ total de tous les
atomes du composé - le nombre de protons total de ce composé (C'est le nombre
d'électrons en plus ou en moins par rapport à la neutralité électrique du
composé).
Le composé passe de l'état d'oxydation n1 à l'état d'oxydation
n2 par une réaction d'oxydation de potentiel V1,2 (avec
n1 le degré d'oxydation du composé le moins oxydé).
Formule de Luther pour trois états d'oxydation:
V1,3 = (n2 - n1)
V1,2+ (n3-n2) V2,3
(n3 - n1)
A noter que les conditions doivent être les mêmes pour les trois réactions d'oxydation.
Potentiel d'oxydo réduction
Le potentiel varie en fonction du pH.
L'eau pure est constituée de H2O principalement, mais aussi de
[OH-] et de [H3O+], les concentrations de ces
deux espèces étant ≤ à 10-2 mole. Le
milieu est dit acide si [H3O-] est élevée. Il
est dit basique ou alcalin si [OH+] est élevée. On trouve
toujours des ions OH+ en milieu acide et des ions
H3O- en milieu basique. On peut donc écrire les
réactions donnant le nombre d'ions H3O- dans des milieux
basiques et inversement avec les ions OH+ en milieu acide. Le pH est
défini comme la concentration active en ions [H3O-],
soit pH = - log a[H3O-] = - log γ[H3O-]. Comme l'activité est
négligée (γ=1), on a donc:
pH = - log [H3O-]
Un électrolyte très acide ou très basique est un bon conducteur du
courant, ce qui permet de diminuer les pertes électriques lors d'une
électrolyse.
Les concentrations en ions [OH+] et en [H3O-]
sont liés par la constante de dissociation Ke de l'eau:
Ke = [OH+] [H3O-] = 10-14 (à 25°C)
Cela signifie que à 25°C, il y aura toujours la même quantité dissocié d'eau dans 1 litre d'eau. Seule la proportion des deux phases dissociées peut varier, faisant ainsi varier le pH. Quand les deux [OH+] et [H3O-] sont égaux, soit 10-7 mole/l, le pH est alors de 7. L'eau est dite pure.
Equilibrer une réaction
l'écrire dans le domaine acide (avec des H3O-) ou dans le domaine basique (avec des OH+). Pour un élément de degré d'oxydation positif, il est plus facile d'écrire dans le domaine acide, et inversement pour un élément de degré d'oxydation négatif (comme le CL+ avec un e+ en plus) dans le domaine basique.
En milieu acide:
Ox + n1 H3O- + n2 e+ ⇌ Red + n3 H2O
Le H3O- fournit un
H- à l'oxydant, et il reste une molécule de
H2O. On regarde combien de
H- il faut donner pour transformer tous les O
(avec éventuellement des OH dans l'Ox) de l'oxydant en eau.
Les deux côtés doivent être équilibrés au niveau des e+
excédentaires (faire un contrôle avec les degrés d'oxydation de chaque
éléments, des deux côtés de la réaction. Ce sont les e+ rajoutés
à la réaction de gauche qui permettent l'équilibrage).
Il faut aussi vérifier que tous les atomes sont équilibrés des deux
côtés.
En milieu basique:
Ox + n1 H2O + n2 e- ⇌ Red + n3 OH+
Il n'y a pas forcément besoin de présence d'eau à gauche.
Etablissement des diagrammes tension-pH
Equation de réduction de l'eau en hydrogène sur le diagramme tension - pH :
VH2O/H2 = - 0.06 pH
Cela veut dire que plus le pH augmente, plus le potentiel de réduction de l'eau en hydrogène diminue, ce qui fait qu'on diminue le risque de dégagement d'hydrogène aux électrodes.
Equation de réduction de l'eau en oxygène sur le diagramme tension - pH :
VH2O/O2 = 1,23 - 0.06 pH
On essayera de travailler dans le domaine de l'eau, car il ne se produit pas de réactions parasites de production d'O2 ou de H2, qui en plus de consommer inutilement du courant produisent des produits inflammables qu'il est difficile de récupérer.
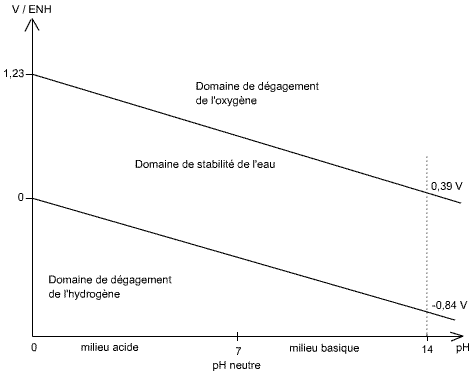
Fig 1 : Diagramme tension-pH de l'eau
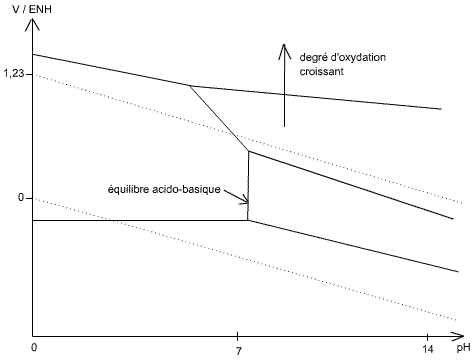
Fig 2 : Diagramme tension-pH du nickel
Conventions prises pour l'établissement du diagramme: (à
reprendre)
convention 1: la concentration totale de toutes les espèces solubles
est inférieure ou égale à 10-2 moles/litres.
convention 2: Les lignes frontières représentent la limite entre deux
espèces, c'est à dire au dela de laquelle l'espèce soluble sera en
concentration inférieure à 10-2 moles/litres.
convention 3: Les rapports des concentrations [base]/[acide] ou
[Ox]/[Red] sont égaux à 1 (cas de deux espèces solides).
convention 4: Dans le cas de présence de gaz.
Méthodologie pour l'établissement du diagramme:
1) Liste de toutes les espèces à considérer.
2) Bilan des espèces dissoutes (ou solubles) : Si une espèce n'est pas neutre électriquement (ex Ni2-, mais pas Ni(OH) qui n'a pas de signe dans l'expression de la molécule, bien que ce soit en fait du Ni- équilibré électriquement par du OH+) elle est soluble. Sinon elle forme un précipité solide de concentration 1.
3) Equilibre acido-basique : (entre 2 espèces de même degré d'oxydation). Donne le pH marquant la limite de présence d'une ou de l'autre espèce. On utilise le Ke et le Ks de la réaction.
4 ) Éléments d'oxydo-réduction : On écrit les réactions entre tous les états d'oxydation possibles.
S'il y a un équilibre acido-basique, de chaque côté de ce pH, le potentiel pour monter d'un état d'oxydation est le même, car sur la courbe c'est un point triple.
Expliquer le fonctionnement du diagramme avant, afin de bien comprendre les frontieres.
Mettre le schéma de montée des différents états d'oxydation.
Diagramme de Frost: Plus un élément est bas dans le diagramme, plus il sera stable.
Double couche:
Voir la partie sur la corrosion.
Cinétique
Une réaction peut être prévue par la thermodynamique vue ci-dessus mais
ne pas se passer de manière préférentielle car sa vitesse de réaction est
trop lente. Ceci est provoquée par la double couche de Helmotz, générant une
surtension η trop grande (à la
cathode cette surtension ηc est très
supérieure à celle de l'anode, ηa)
du à un mauvais couple matériau électrode/électrolyte.
Plusieurs solutions:
- Changer le matériau de l'électrode
- Changer l'électrolyte en y mettant un complexeur ou un inhibiteur
- Changer les conditions de travail (agitation, température, ...)
Générateur
Tension U disponible aux bornes d'un générateur:
U= Veqfem - I.ΣR - ηa -
ηc
avec I le courant débité, et I.ΣR - ηa - ηc les pertes. Ces pertes
sont positives dans le cas de la recharge. ηa
<< ηc, la surtension étant fonction du couple
matériau de l'électrode/électrolyte.
à suivre...